基礎軟件服務 數字化轉型的堅實基石
在數字經濟蓬勃發展的今天,基礎軟件服務作為信息技術產業鏈的底層支撐,其重要性日益凸顯。它不僅是企業數字化轉型的關鍵驅動力,更是構建安全、高效、智能現代信息社會的核心要素。
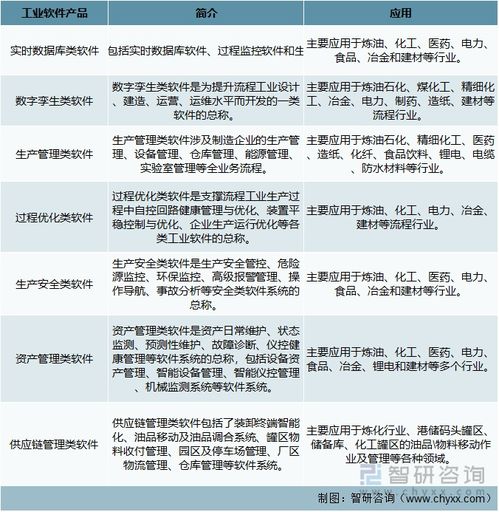
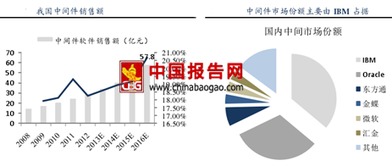
基礎軟件服務通常指操作系統、數據庫、中間件等為上層應用提供通用性、基礎性功能支持的軟件平臺與服務。與直接面向終端用戶的應用軟件不同,基礎軟件如同信息世界的“地基”與“骨架”,默默承載著各類業務系統的穩定運行。例如,操作系統管理著硬件資源,為所有應用提供統一的運行環境;數據庫系統則負責海量數據的存儲、管理與高效訪問,是數據價值挖掘的前提。
當前,基礎軟件服務領域呈現出顯著的發展趨勢。是云化與服務化。傳統授權模式正在向訂閱制、SaaS(軟件即服務)模式轉變,用戶無需關注復雜的部署與維護,可按需獲取彈性、可擴展的計算、存儲與數據服務,大大降低了技術門檻與總體擁有成本。開源生態持續繁榮。以Linux、Kubernetes、MySQL等為代表的開源項目,通過全球協作創新,已成為基礎軟件領域的重要力量,推動了技術的快速迭代和行業標準的形成。自主可控與安全成為重中之重。在全球技術競爭與網絡安全威脅加劇的背景下,發展安全可靠、具備自主知識產權的基礎軟件,保障關鍵信息基礎設施供應鏈安全,已成為國家與企業的戰略共識。
從價值維度看,優質的基礎軟件服務為企業帶來多重收益。它能夠提升IT系統的穩定性與性能,保障業務連續性;通過標準化和自動化,提升開發和運維效率,加速產品上市時間;其開放性與兼容性也有助于企業集成新舊系統,打破信息孤島,構建靈活可擴展的IT架構。
隨著人工智能、物聯網、邊緣計算等技術的深度融合,基礎軟件服務將向著更加智能化、泛在化、一體化的方向演進。例如,數據庫將內置更多AI能力以進行自治優化;操作系統需更好地適配從云端到邊緣的多樣化算力場景。
總而言之,基礎軟件服務雖處“幕后”,卻至關重要。它既是技術創新的沃土,也是產業升級的引擎。持續夯實這一基石,對于個人、企業乃至國家在數字化浪潮中把握主動權、贏得未來競爭力,具有不可替代的戰略意義。
如若轉載,請注明出處:http://www.ml7tnu00.cn/product/50.html
更新時間:2026-06-19 17:12:12